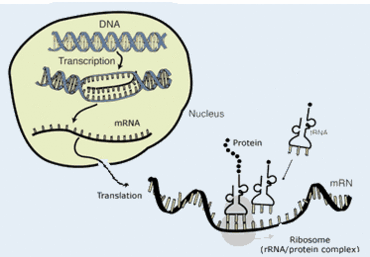
AKI (Acute Kidney Injury) is a common occurrence for ICU (Intensive Care Unit) patients and is associated with increased mortality and complication rates. In such cases, renal replacement therapy (RRT) is needed to ensure patient survival. Predicting outcomes of RRT is therefore key to identifying patients at increased risk for mortality and/or end-stage renal disease (ESRD), as well as those that will likely best respond to therapy.
To this extent, influence factors such as RRT modality, time of RRT initiation and/or cessation, treatment parameters, comorbidities, ICU scores, lab values and urine output shall be analyzed. In particular, patient outcomes of interest are, among others, mortality (90-day and in-hospital), time for the recovery of renal function, development of ESRD, hemodynamic stability and length of ICU stay.
Your task therefore is to develop and evaluate a clinical prediction model (CPM) based on factors mediating patient outcomes following RRT in the ICU by means of machine learning methods. To achieve this purpose, you will utilize established ML toolkits and have the opportunity to go through the whole process of developing a CPM: data extraction, feature engineering, algorithm selection, model development and validation. A model thus developed can be used as for foundation for a Clinical Decision Support System to aid in the therapy planning of kidney patients.