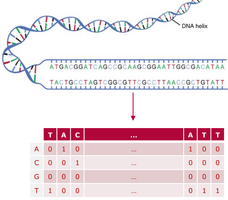
Genomic sequences are long strings with one of the 4 nucleotides (Adenine, Guanine, Cytosine, Thymine) at each position. To obtain a numeric representation, sequences are one-hot-encoded resulting in a matrix with the size sequence length x 4. These matrices can be then used as input for the network.
CNN's main architecture is made of two different kinds of neural network layers.
1. Convolutional layers consist of matrices (so-called filters or kernels) which learn local representations of patterns within the data that are relevant for the prediction task. In the case of genomic sequences, convolutional layers learn (sub-) motifs. Depending on the networks architecture, motifs are learned directly in the first layer or distributed among deeper layers.